Feature Correspondence#
This section describes the theoretical background for the LC-MS feature matching algorithm,
implemented in the LCFeatureMatcher operator.
Feature correspondence is a necessary step before performing any kind of statistical comparison between samples. Smith et al [1] discuss in detail the different methods and issues in feature correspondence in LC-MS. The algorithm developed for TidyMS2 tries to address these issues. In this section we describe the algorithm that is used for performing feature correspondence, and that is a slightly modified version of the original algorithm described in the TidyMS paper [2].
More specifically, feature correspondence refers to group features across samples based on their chemical identity. We refer to such groups as feature groups.
In principle, only one feature from each sample must be included in a given feature group. This is not strictly true and it will be discussed in more detail in this guide, but for a first approximation to the feature matching process, we will assume that this is a requirement.
In TidyMS2, a cluster-based approach is to build feature groups, using the feature descriptors from all samples as input. Features that are close in terms of m/z and Rt are associated to the same feature group.
To avoid having more than one feature from a sample in a given feature group, a multi-step strategy is used: first, the DBSCAN algorithm is used to group features based on spatial closeness using Rt and m/z. In a second step, the number of ionic species on each cluster created by DBSCAN is estimated based on the number of features per sample in the cluster. Subsequently, clusters for containing a single ionic species are built by using a Gaussian Mixture Model (GMM) with a number of components equal to the number of ionic species estimated in the previous step. Using the GMM model, features in a sample are assigned to a ionic species in an unique way by solving an assignment problem. In a final step, clusters evaluated and merged if they are associated with the same chemical species. We now describe each step in detail.
DBSCAN clustering#
For the initial clustering step, the scikit-learn implementation of the DBSCAN algorithm is
used. DBSCAN is a non-parametric, widely used clustering algorithm that build clusters connecting
points that are closer than a specified distance eps. It classifies points according to the
number of neighbors that they are connected to: core, if they are connected to min_samples
or more points including itself, reachable if they are connected to a core point, and noise
otherwise. eps is set based on the experimental precision of the mass spectrometer and the
dispersion expected on the Rt based on the separation method used. We found that using two times
the maximum expected standard deviation for Rt and m/z produces the best results. See
this section for a description of the method used to select defaults
values.
The rt_tolerance and mz_tolerance parameters of the feature matching function are defined
to account for variations in m/z and Rt. A value of 0.01 for mz_tolerance
is used for Q-TOF instruments and 0.005 is used for Orbitrap instruments. In the case of
rt_tolerance, 5 is used for UPLC and 10 for HPLC (in seconds).
The Rt feature descriptor is scaled using these parameters:
rt_scaled = rt * eps_mz / eps_rt. In this way, mz_tolerance can be used as the eps
parameter of DBSCAN. The min_samples parameter of the DBSCAN model is computed from the
min_fraction parameter using the minimum number of samples in the included classes:
min_samples = round(min_fraction * n), where n is the smallest number of samples in a
class in include_classes. Since the dispersion is Rt and m/z is independent, the distance
function used is the Chebyshev distance.
As mentioned before, the feature clusters created in this way may contain more than one chemical
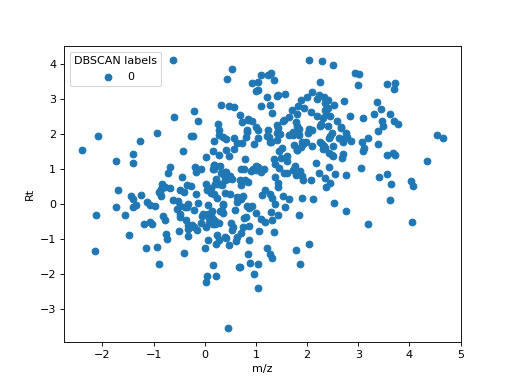
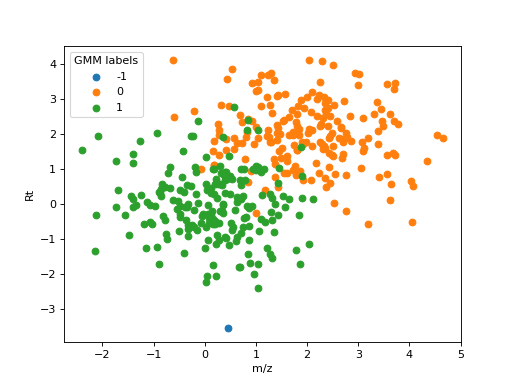
species. As an example, the following figure shows an example of clustering two close ionic
species using DBSCAN. 200 observations of samples with distribution \(\sim N(0, 1)\) and
\(\sim N(1, 1)\) in m/z and Rt were used to simulate two ionic species with close values
detected in 200 samples. Using eps=2 and min_samples=50, all features were grouped
together in a single cluster.
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
DBSCAN clustering applied to two close ionic species.#
This example shows why the subsequent steps in the feature matching algorithm are required.
Assigning features to ionic species#
After clustering features using DBSCAN, the number of ionic species in each cluster is estimated: the number of features from each sample is counted and used to define the k-feature repetitions \(n_{k}^{\textrm{(rep)}}\) in a cluster, that is, the number of times a sample contribute with k features to the cluster. For example, if in a cluster the number of samples that contribute with two features to the cluster is 20 then \(n_{2}^{\textrm{(rep)}}=20\). The number of ionic species \(n_{s}\) in a cluster is defined as follows:
where \(n_{\textrm{min}}\) is the parameter min_samples computed for DBSCAN. \(n_{s}\)
is used to set the n_components parameter in a GMM, trained with all the features found in
the cluster. After training the GMM, a matrix \(S\) with shape \((n_{c} \times n_{s})\) is
built for each sample, where \(n_{c}\) is the number of features that the sample contributes
to the cluster. \(s_{ij}\) is defined as follows:
Where \(mz_{i}\) and \(rt_{i}\) are the m/z and Rt values for the i-th feature,
\(\mu_{mz, j}\) and \(\mu_{rt, j}\) are the means in m/z and Rt for the j-th ionic species
(j-th component of the GMM) and \(\sigma_{mz, j}\) and \(\sigma_{rt, j}\) are the standard
deviations for the j-th ionic species. S can be seen as a measure of the distance to the mean of
each cluster in units of standard deviations. Using \(S\) we can assign each feature to an
ionic species in a unique way using the Hungarian algorithm [3]. If \(n_{c} > n_{s}\),
features that were not assigned to any ionic species are assigned as noise. After all features in
a sample are assigned, the value of \(s_{ij}\) is checked. If it is greater than
max_deviations, which is set to 3.0 by default, the feature is assigned to noise. In the
context of feature correspondence, assigning a feature to noise means that the feature does not
belong to any feature group and will not be included in the data matrix.
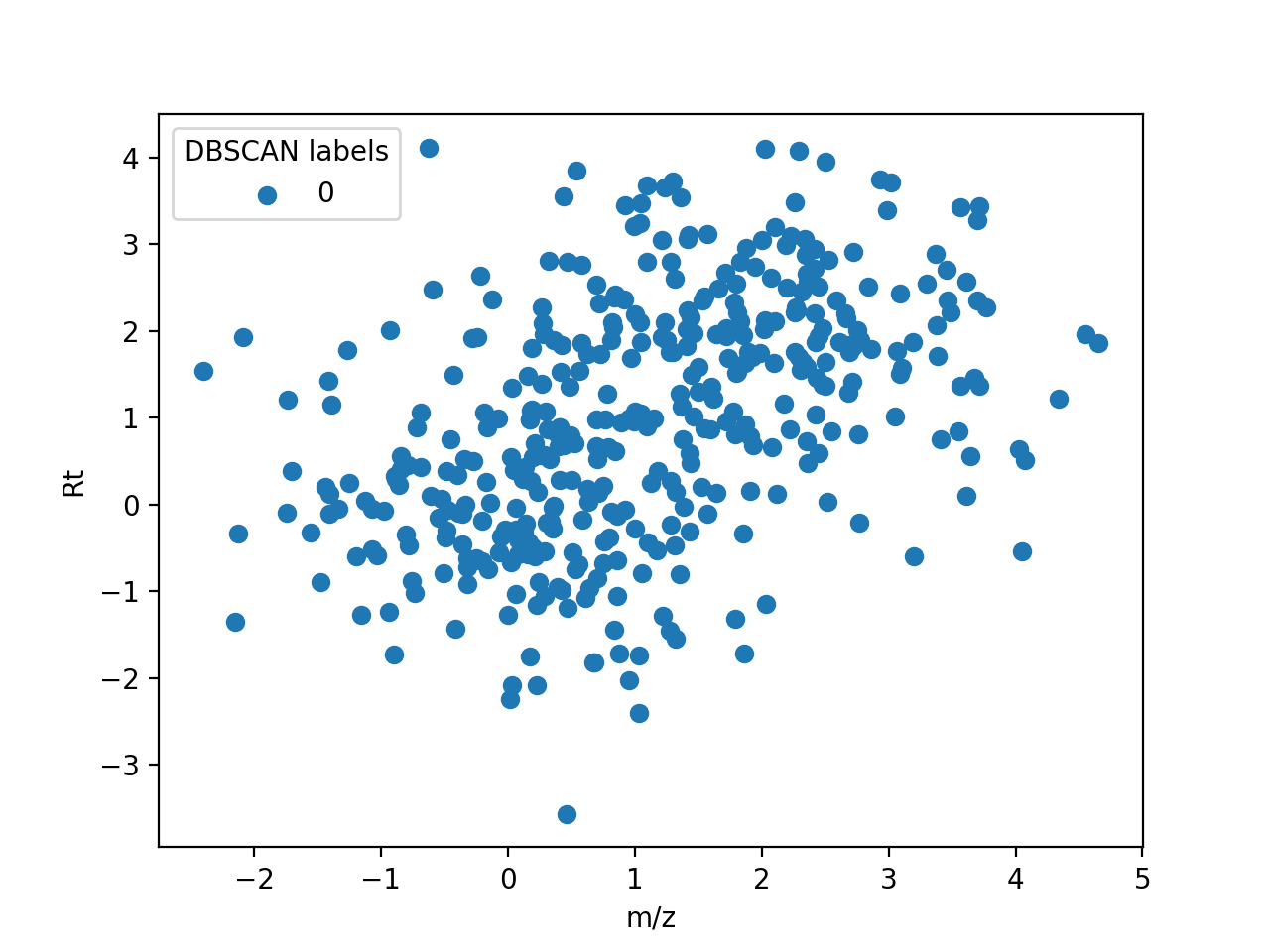
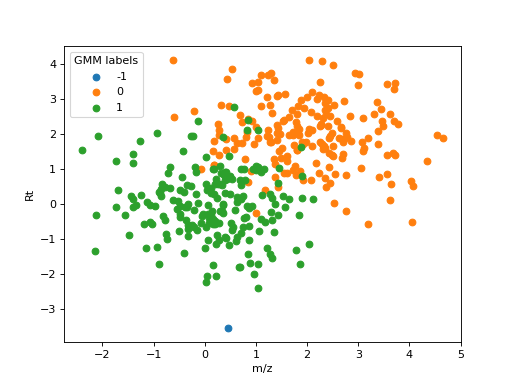
The following figure shows how each feature in the example shown for DBSCAN is assigned to a unique ionic species:
(Source code, png, hires.png, pdf)
{kind=link}
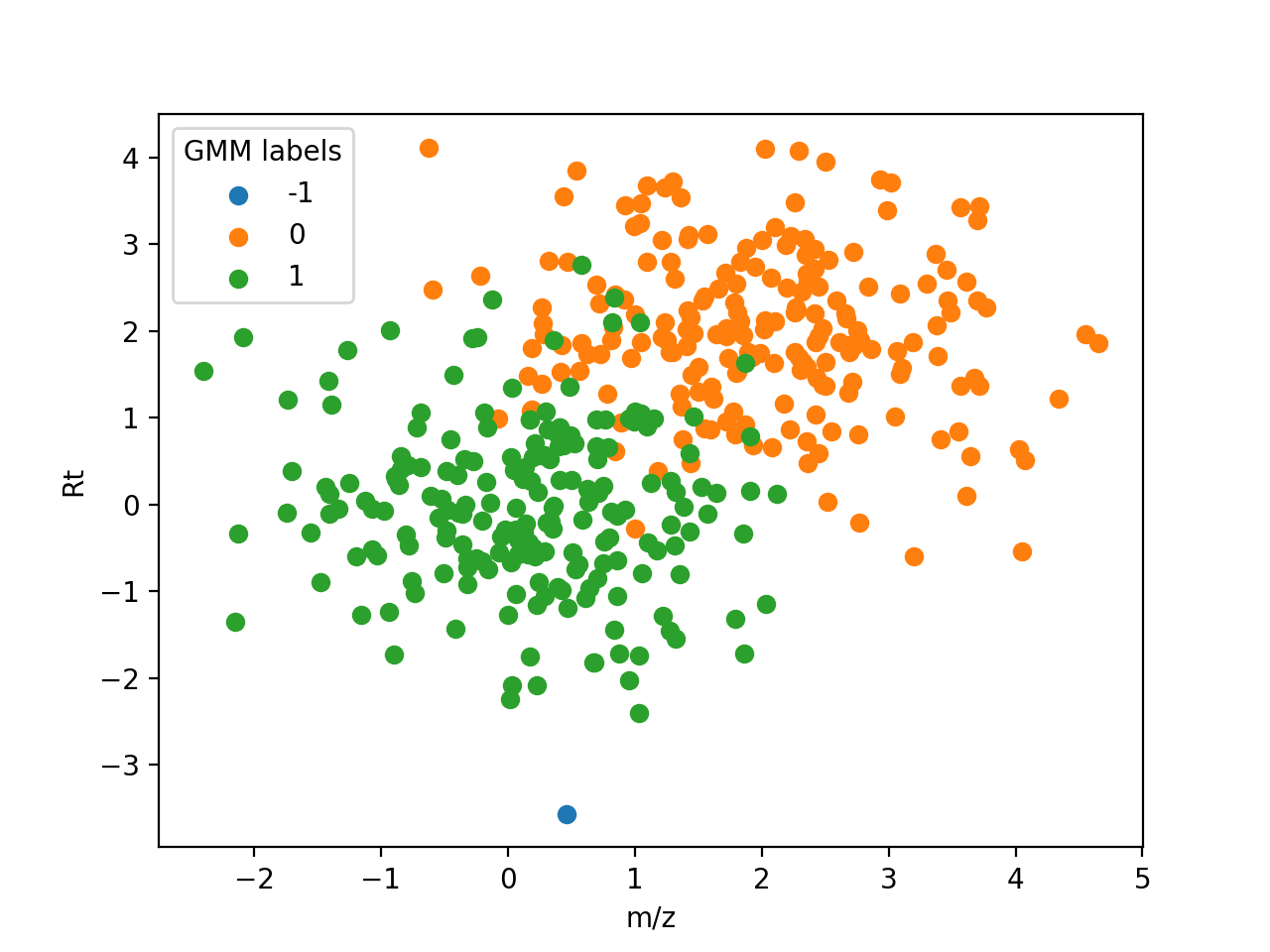{kind=link}
Assignment of features to a unique ionic species. Features labelled with -1 are noise.#
Merging GMM clusters#
Up to this point we required that feature clusters contain at most one feature from each sample. There are some cases in which it may be useful to create clusters with more than one feature from one sample. This may happen for multiple reasons. One of these reasons is that features m/z and Rt are so close that the instrument resolution is not enough to distinguish them as two separate entities in all samples. This results in detecting both features in some samples, and a combination of the two in a single feature in other samples.
To solve this issue, the max_overlap parameter is included. This parameter compares pairs
of close feature clusters and merge them if their overlap is lower than the max_overlap.
A pair of features is considered close if the distance between their centroids both in m/z
and Rt is lower than their respective tolerance parameters. The overlap is defined as
the fraction where both clusters contain a feature from the same sample. This simple mechanism
allows to check distinguish cases where the instrument resolution was enough to discriminate
the pair of features in a significant number of samples. By default, max_overlap is set to
0.25, i.e., it must be possible to distinguish the features in at least the 25 % of samples.
There is also another scenario that benefits from cluster merging and it is when features have different mean values in different subsets of samples, something that results in the creation on multiple clusters for the same chemical species.
Default values for the DBSCAN parameters#
The main goal of the application of the DBSCAN algorithm is to cluster features
from the same ionic species. One of the assumptions is that the values of Rt
and m/z in a ionic species are randomly distributed around its true value. Also,
before training the DBSCAN model, Rt values are scaled using eps_rt and
eps_mz, which are greater than the maximum expected dispersion for m/z and
Rt. After this step, the standard deviation in Rt should be equal or lower than
the standard deviation in m/z. It is for this reason that the analysis can be
limited to cases where the standard deviation in Rt and m/z are the same. For
the evaluation of the DBSCAN parameters we simulate m/z and Rt values using
the standard Normal distribution.
The effect of different parameters are tested using different sample sizes,
and repeating each test five times. The following values were tested:
min_sample: 10 %, 25 %, 50 %, 75 % and 100 % of the current sample size.eps: 0.5, 1, 2, 3 and 4.
To measure the performance to cluster the data the noise fraction was evaluated, defined as the ratio between the number of samples classified as noise and the total number of samples. The following figure shows the result from this analysis.
(Source code, png, hires.png, pdf)
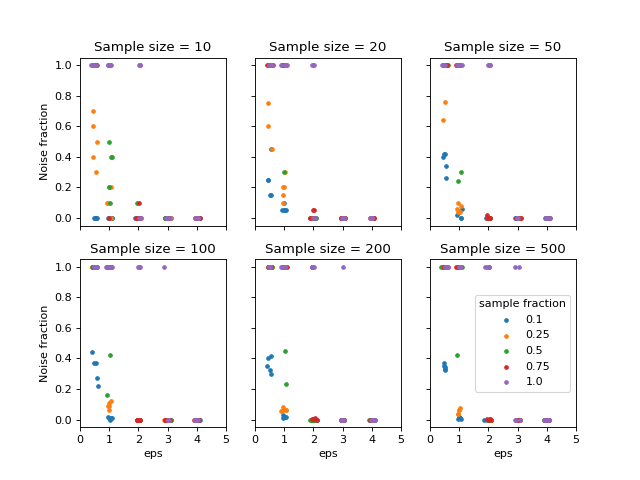{kind=link}
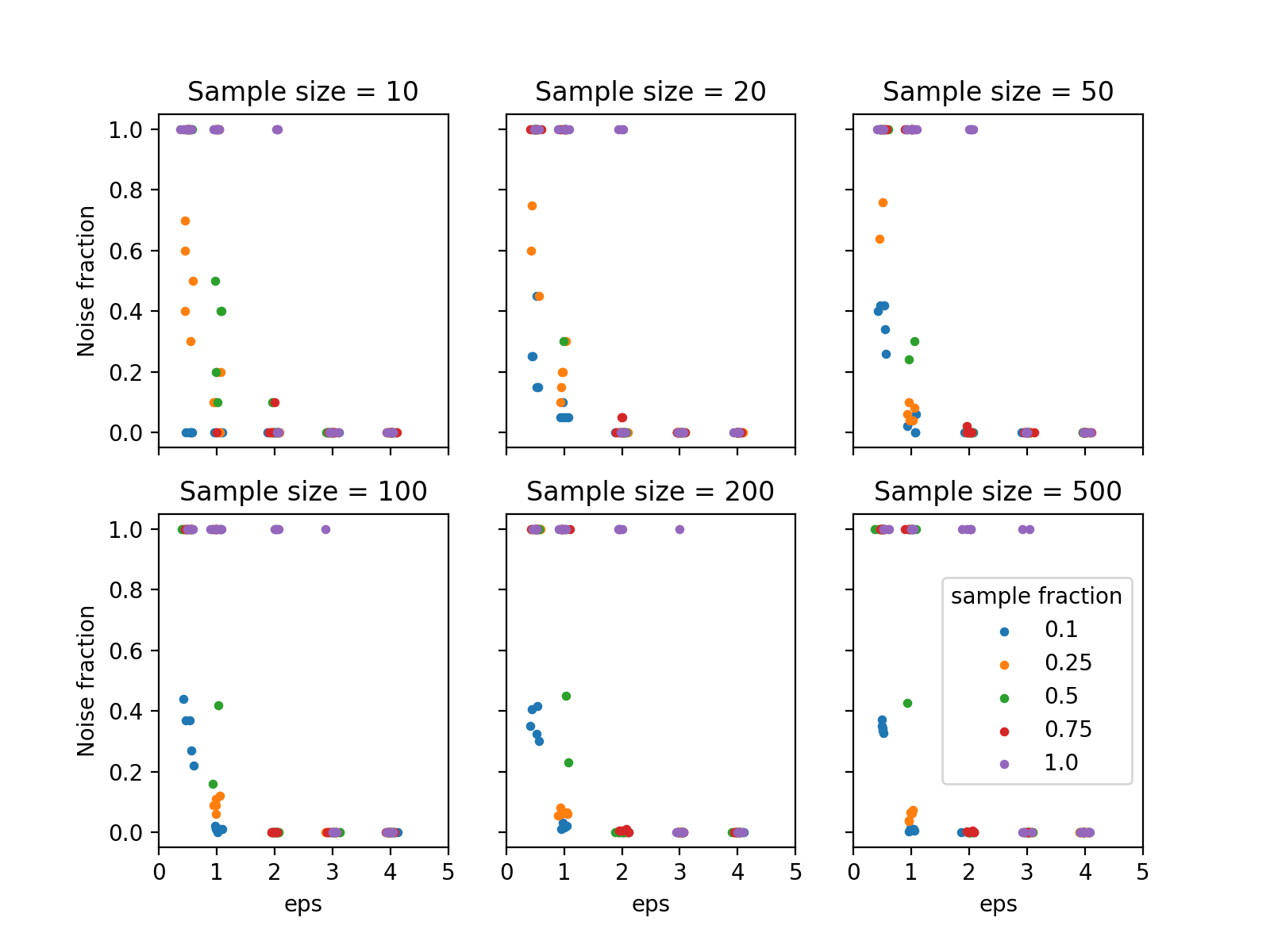{kind=link}
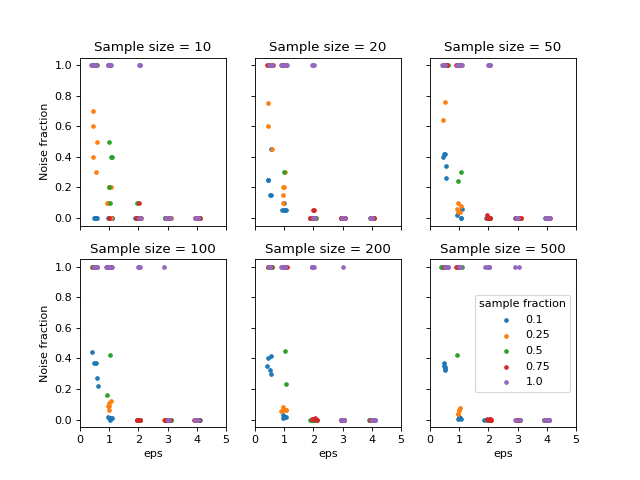
Noise fraction for different parameters used in DBSCAN.#
It can be seen that eps >= 2 and min_samples <= 0.75 * n reduces the
noise fraction to zero in almost all cases. Based on this, eps=2.0 and
min_samples=0.25 * n seem a reasonable choice. The next step is to translate
the value of eps to eps_mz and eps_rt. In the case of eps_mz,
the values are computed from the experimental deviation commonly observed
according to the instrument used. For example, for Q-Tof instruments standard
deviations of 3-4 mDa are common. Based on this, the default value is set as
0.01. In the case of eps_rt the election of a default value is not
so straightforward. We choose a default value for UPLC of 5 s based on the
typical values observed on experimental data.